THALASSEMIA-TYPES & PATHOPHYSIOLOGY
- What are the types of mutations of beta Thalassemia?
Beta thalassemia is
a spectrum of disrorder characterised by variable clinical and laboratory
features that occur due to varying degree of beta gene defect resulting into
defect in eta globin synthesis.
The beta globin
synthesis may be completely absent or it may be reduced or it may be present
with other disorders of Hb synthesis.
Depending upon the
abnormality of globin synthesis beta mutation can be
· β+=
reduced beta globin synthesis
· β0=complete
absence of beta globin synthesis
Since
2 beta genes are present one in each chromosome the mutation in two genes will
determine the clinical presentation.


Both these conditions
cause
TRANSFUSION DEPENDENT BETA THALASSEMIA MAJOR.
Severe anemia, dependent
on regular blood transfusion for survival
HbF elevated.
PATHOPHYSIOLOGY


2 What are the determinants of severity of β-thalassemia?
The major determinant of severity is the ratio of alpha
and non-alpha globin chains.
The degree
of non-alpha (i.e. beta in this case) production depends upon the type of mutation(β+ or β0 ) and zygosity (β+/ β0 or β0/β0
or β+/β+
).
3 What are the other modifiers of disease
severity?
i) Concomitant alpha
thalassemia-
Reduces disease
severity
Because the ratio of
alpha and beta is partially normalized
ii) Increased HbF
Reduces severity as gamma chains will be
available to form tetramer with alpha chains.
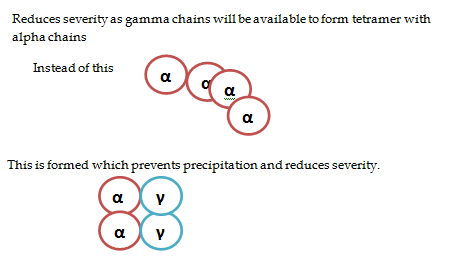
iii) Sickle-beta thalassemia –
iv) Co-inheritance of hemoglobin E –
Hemoglobin E results from a beta globin mutation that reduces beta globin production. The clinical phenotype is similar to beta thalassemia but less severe. Hemoglobin E is commonly seen in India and Southeast Asia.
Co-inheritance of heterozygosity for a beta thalassemia mutation and a hemoglobin E mutation is responsible for a large proportion of severe beta thalassemia throughout the world.
The phenotype appears to be especially severe in Sri Lanka . The pathophysiology of this heterogeneity and its severity remain a puzzle. Co-inheritance of alpha thalassemia appears to have a significant impact.
4. What is HPFH?
Hereditary persistemce of
fetal hemoglobin.
It is due to absence of
one or both delta or beta chains with resulting increase gamma chains leading
to increased HbF.
Most of the cases have no
anemia with increased HbF levels in electrophoresis.
They can get co-inherited
with other defects like sickle cell where they can reduce the severity of
sickle cell disease.

5. What leads to the ineffective erythropoiesis /intramedullary hemolysis?
In thalassemia the RBCs formed containing abnormal HB
gets destroyed within the BM thus k/a intramedullary hemolysis.
Thus the premature RBC death caused by maturation
arrest followed by apoptosis due to the toxic alpha globin chain causes ineffective
erythropiesis.
6. Why is there accelerated hemolysis in Thalassemia?
Hemolysis is Thalassemia is primarily intracorpuscular.
The causes of accelerated
RBCs derstruction in THALASSEMIA are:
·
The unstable alpha
globin peptides which appear as inclusions in the RBC membrane which makes RBC
memebrane unstable.
· The RBC membrane composed of Band 3, Band 4.1 rotein,ankyrin and spectrin are destabilized.
· The oxidative stress is high.
·
Also the
thalassemic RBCs have low Hb and are dehydrated.
7. Why is there high oxidative stress in RBCs of thalassemic patients?
Oxidation of Hb occurs in all the RBCs in normal individual forming small fraction of methemoglobin which gets reduced back to Hb by cytochrome b5 reductase.
In thalassemia oxidation of alpha and beta chain
produce HEMICHROMES which cannot get reduced back to normal Hb.
These iron conatining hemichromes can produce reactive oxygen species thus damaging the proteins and lipids of RBC membrane.
8. Why are the RBCs of thalassemia patient dehydrated?
Due to excessive activation of k and Cl channel which draws
water along with it extracellularly.
Thus MCHC is high in Thalassemia.

9. There is found to increased risk of thrombosis in Thalassemia patients may be due to increased expression of phosphotidylserine C in the RBC membrane.
10.Increased risk of infection with Yersinia enterocolitica.
11. What are the types of alpha thalassemia?


12. Why is alpha thalassemia less severe than beta thalassemia?
In the alpha thalassemias, the excess beta or gamma globin chains can
form partially soluble but ineffective hemoglobin homotetramers.
These homotetramers do not precipitate extensively until they are
exposed to damaging effects in the circulation, mostly oxidant in nature.
However the HB barts i.e. Hydrops fetalis is imcompatible with life.
13. What is hemoglobin constant spring?
It is a non deletional type of alpha thalassemia where
the genes are not deleted but mutated.
It is commonly associated with HBH disease.
In contrast to beta thalassemia alpha thalassemia are hyperhydrated.
The Hb constant spring produce more severe anemia and more severe disease.
14. What is hemoglobin Lepore?
A fusion globin due to unequal crossover of the β- and
δ-globin genes (the globin is produced at a low level because it is under
δ-globin regulation).
These patients are asymptomatic.
15. Anti –Lepore also results from opposite crossover between the β- and δ-globin genes.
16. Hb Kenya — Hb Kenya is an interesting, clinically innocent hemoglobinopathy that results from an unequal crossover and recombination event between the beta and A(gamma) globin genes
17. Why is there iron overload in the patients of thalassemia even before the transfusion?
1.



Comments
Post a Comment